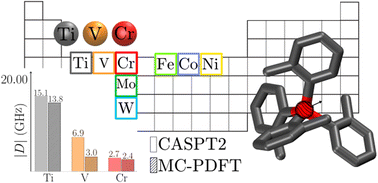
In the realm of optically addressable qubits, a previously synthesized and characterized Cr(IV) pseudo-tetrahedral complex, featuring four strongly donating ligands surrounding the chromium center, has demonstrated potential as a qubit candidate. This study proposes analogs of this complex through a metal substitution strategy, extending the investigation to different complexes based on metal centers selected from first-row and Group 6 transition metals. Computational modeling based on multiconfigurational methods CASPT2 and MC-PDFT was utilized to calculate energy gaps between ground and excited electronic spin states, and zero-field splitting parameters. Simulations were applied to each equilibrium geometry and related deformations based on vibrational modes. All results align with previous experimental findings, but also show that qubits based on V and Ti centers could be more electronically stable than the Cr one, suggesting a lower electronic features dependency from their related geometry. In some cases geometrical deformations provide changes in relative energy gaps between triplet and singlet excited state, that could potentially swap, offering a different initialization process, and some inspiration for ligand design based on such deformations. Additionally, this study identifies an unsynthesized Ti(II) compound as a promising candidate for molecular qubits. This finding highlights the role of computational multireference methods in the rational design of qubit systems.

